单细胞分析工具—scCODA 与scCODE

scCODA——细胞组成比较
scCODA(single-cell compositional data analysis)是由德国环境健康研究中心计算生物学研究所M Büttner等人基于python开发的单细胞数据分析工具,于2021年11月发表于Nature Communication;主要用于分析不同分组样本的细胞组成的差异。参考官方文档记录用法如下。
Paper:/articles/s41467-021-27150-6Github:https://github.com/theislab/scCODATutorial:/en/latest/index.html
1、安装环境
conda create -n sccoda python=3.9conda activate sccoda
conda install rpy2
pip install sccoda
# conda install -c conda-forge notebook
2、分析流程
(1)加载函数
import importlib
import warnings
warnings.filterwarnings("ignore")
import pandas as pd
import pickle as pkl
import matplotlib.pyplot as plt
from sccoda.util import comp_ana as mod
from sccoda.util import cell_composition_data as dat
from sccoda.util import data_visualization as viz
import sccoda.datasets as scd
(2)读取数据
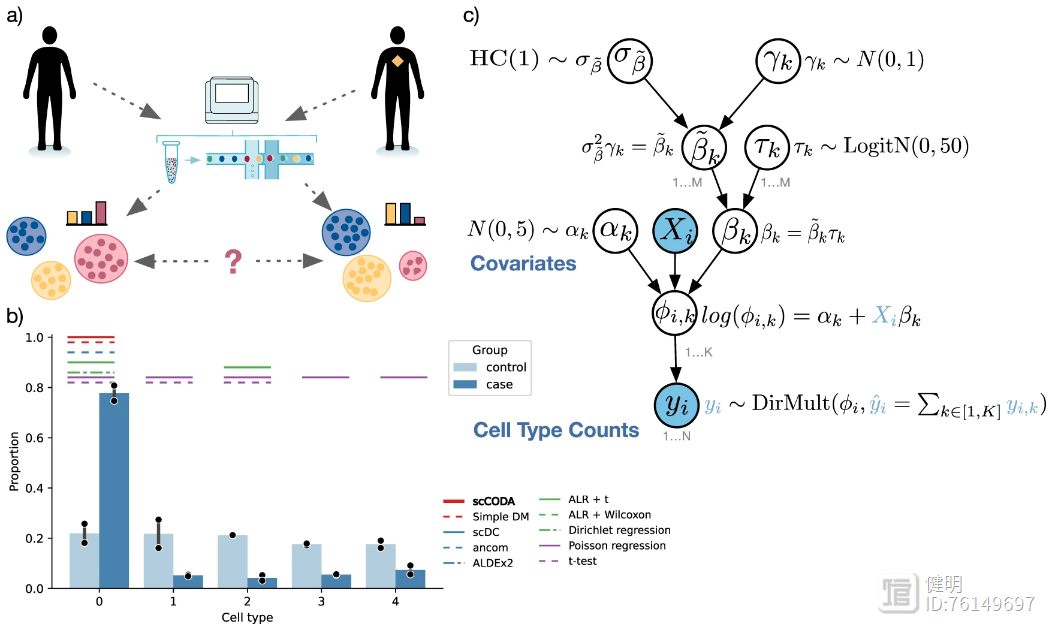
pandas.Dataframe:第一列为样本名,其余每列各代表一种细胞类型,值表示细胞数量使用scanny包转换为Anndata结构格式,obs表示样本信息## 导入示例数据
cell_counts = scd.haber()
print(cell_counts)
#
Mouse Endocrine Enterocyte Enterocyte.Progenitor Goblet Stem TA TA.Early Tuft
# 0
Control_1
36
59
136
36 239 125
191
18
# 1
Control_2
5
46
23
20
50 11
40
5
# 2
Control_3
45
98
188
124 250 155
365
33
data_all = dat.from_pandas(cell_counts, covariate_columns=["Mouse"])
data_all.obs
data_all.X
## 提取分组信息
data_all.obs["Condition"] = data_all.obs["Mouse"].str.replace(r"_[0-9]", "", regex=True)
#
Mouse
Condition
# 0
Control_1
Control
# 1
Control_2
Control
# 2
Control_3
Control
data_salm = data_all[data_all.obs["Condition"].isin(["Control", "Salm"])]
(3)组成差异分析
## 设置先验信息
model_salm = mod.CompositionalAnalysis(data_salm,
formula="Condition", #指定参考组
reference_cell_type="Goblet") #指定一种细胞类型作为已知组间比例不变的标准,如不确定,则可以设置为 "automatic"
## Markov-chain Monte Carlo (MCMC) inferrence
sim_results = model_salm.sample_hmc() # time consuming
# 备选方案:sample_hmc_da(), sample_nuts()
## 分析结果
# sim_results.set_fdr(est_fdr=0.4)
sim_results.summary()
sim_results.credible_effects()
sim_results.effect_df
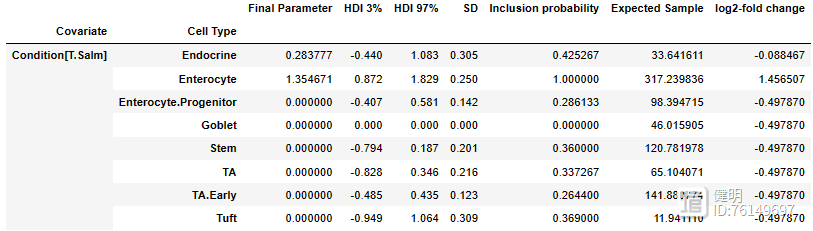
如上重点关注effect的Final Parameter列
若为0表示,该细胞类型比例在组间差异不大,可设置set_fdr设置判断的阈值标准大于0,则表示相对于参考组,细胞比例提高;反之,则相反。
3、可视化
此外scCODA也提供了一些可视化细胞比例的绘图函数,简单示例如下。
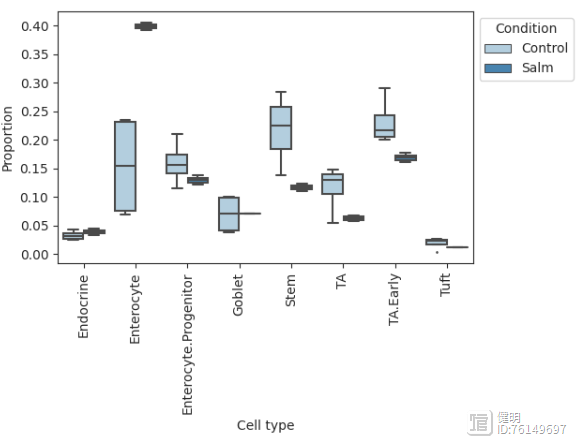
箱图viz.boxplots(data_salm, feature_name="Condition")
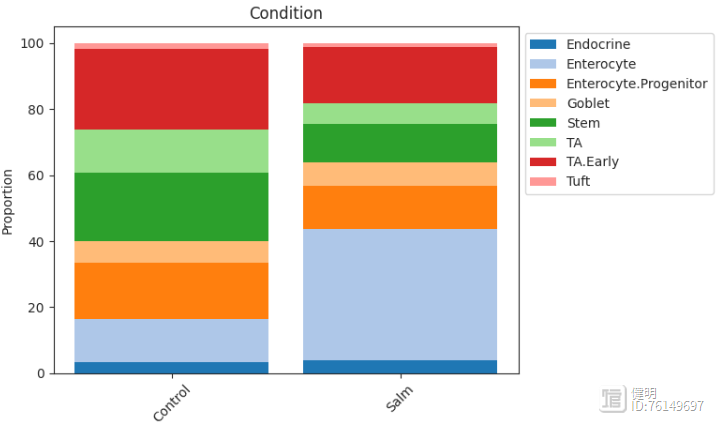
柱状图viz.stacked_barplot(data_salm, feature_name="Condition")
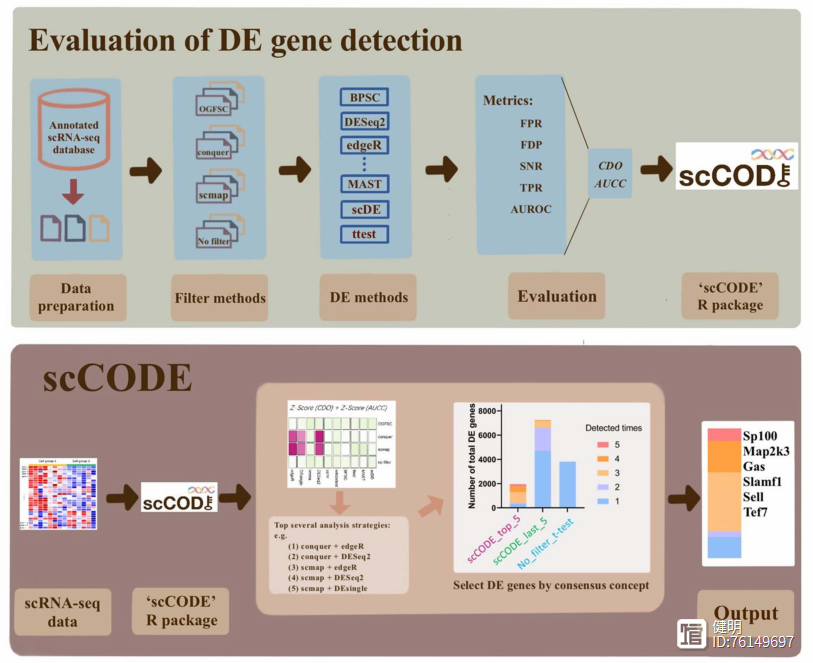
scCODE综合差异分析
scCODE( single-cell consensus optimization of differentially expressed gene detection)是由复旦大学附属金山医院邹欣等人开发的R包工具,于2022年12月发表于Briefing in Bioinformatics;该工具对多种差异基因分析策略进行了集成、整合,用于鉴定鲁棒性的单细胞差异基因。用法比较简单,简单记录如下。
Paper:/bib/article-abstract/23/5/bbac180/6590434?redirectedFrom=fulltextGithub:https://github.com/XZouProjects/scCODE
1、安装R包
necessary1 <- c('doParallel', 'samr','doSNOW','pls')installed <- necessary1 %in% installed.packages()[, 'Package']
if (length(necessary1[!installed]) >=1){
install.packages(necessary1[!installed])
}
necessary2<-c('DESeq2', 'DEsingle',
'edgeR', 'limma', 'MAST', 'S4Vectors', 'scDD', 'scmap', 'SingleCellExperiment', 'SummarizedExperiment')
installed <- necessary2 %in% installed.packages()[, 'Package']
if (length(necessary2[!installed]) >=1){
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
library(BiocManager)
BiocManager::install(necessary2[!installed])
}
install.packages("BPSC_0.99.2.tar.gz", repos = NULL, type="source")
install.packages("OGFSC_0.2.3.tar.gz", repos = NULL, type="source")
install.packages("scCODE_1.2.0.0.tar.gz", repos = NULL, type="source")
2、差异基因分析
(1)准备两组单细胞样本的count表达矩阵
library(scCODE)
data1<-data1_sccode
data1[1:4,1:4]
#
[,1] [,2] [,3]
[,4]
# Gnai3 12336.737462
0
0 5399.62
# Cdc45
0.000000
0
0
0.00
# Narf
0.000000
0
0
0.00
# Scmh1
8.639172
0
0
0.00
dim(data1)
# [1] 13045 139
data2<-data2_sccode
dim(data2)
# [1] 13045 323
(2)差异分析
默认light模式下,使用5种策略进行分析;再统计每种策略的判断结果。如果一个基因的5种结果均判断为显著差异基因,则相对更可靠。在linux端使用时,出现类似OpenBLAS blas_thread_init: pthread_create failed for thread 60 of 128: Resource temporarily unavailable报错,经查在shell命令行设置如下参数可正常使用。 export OPENBLAS_NUM_THREADS=2
export GOTO_NUM_THREADS=2
export OMP_NUM_THREADS=2
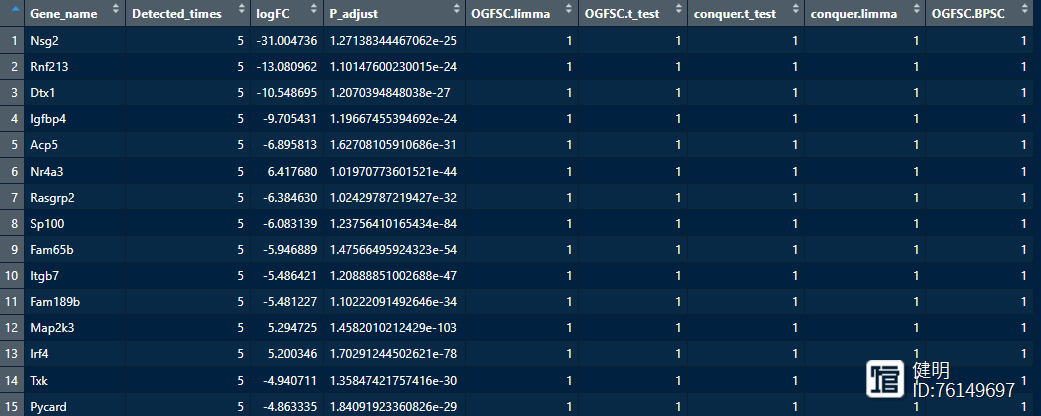
results<-scCODE(data1,data2,light = TRUE,top_ranked=5)
deg = results$DE_results
table(deg$Detected_times)
# 1
2
3
4
5
# 360 1287 132 496 917
head(deg)
Photoshop作为最常用的工具之一,提供了多种磨皮处理方法。其中,通道磨皮法是一种非常常用且有效的方式。
Photoshop作为最常用的工具之一,提供了多种磨皮处理方法。其中,通道磨皮法是一种非常常用且有效的方式。随着社交媒体的兴起和人们对美的追求,磨皮处理成为了美女们追求完美肌肤的一种方式。而在数字图像处理软件中,Photoshop作为最常用的工具之一,提供了多种磨皮处理方法。站长网2023-07-29 17:15:300002如何建设养老服务智能化,智慧养老建设
随着我国老龄化进程的不断加快,老年人对养老服务的需求也日益增长。如何为老年人提供更加便捷的养老服务,已成为摆在我们面前的一道难题。在国家政策的支持下,智能养老服务已经开始进入千家万户,它不仅满足了老年人对于日常生活的需求,也大大提高了生活质量。目前的智慧养老程度站长网2023-07-28 09:50:4000005个“学习”网站,是真的!
hello大家好,这里是日常爆肝更新的老Y工作室。周末本想休息,后台有朋友让推荐几个免费学习的平台,其实老Y第一反应是给她推荐B站,里面确实有许多免费的视频。老Y记得前几天有个新闻,B站上播放时长最长的内容是高等数学,可见这届网友还是很爱学习的。老Y现在有啥想知道的也会经常搜索小破站今天再给大家总结5个不错的免费学习平台,里面同样是汇聚了很多免费课程,爱学习的朋友快收藏起来吧。站长网2023-07-27 17:30:230000别盯着CTRL E了,CTRL Q也担得起万能快捷键的称号 -80
普通的快捷键一键一功能。CTRLQ,不普通,它能一对多。当你按下后还需要再次点选需要的功能。有哪些功能?不如亲自试试吧!添加数据条框选目标区域→【CTRLQ】→【格式化】→【数据条】按单元格中的数字自动添加数据条,直观感受数据大小。添加色阶框选目标区域→【CTRLQ】→【格式化】→【色阶】数字越大颜色越浅图标集框选目标区域→【CTRLQ】→【格式化】→【图标集】站长网2023-07-29 13:00:110000